
Translate this page into:
Duodenal paraganglioma - A case report
*Corresponding author: Abirami Manivannan, Department of General Surgery, Sri Ramachandra Institute of Higher Education and Research, Chennai, Tamil Nadu, India. dr.abiramimanivannan@gmail.com
-
Received: ,
Accepted: ,
How to cite this article: Manivannan A, Raja Senthil V, Chandru R, Singh KB. Duodenal paraganglioma - A case report. Sri Ramachandra J Health Sci. 2023;3:76-8. doi: 10.25259/SRJHS_20_2023
Abstract
Gangliocytic paraganglioma (GP) is a rare tumor of the duodenum. Worldwide, less than 300 cases have been documented. According to cases previously reported, the tumor is frequently seen in the second part of the duodenum. In this article, we discuss the case of a young adult who had a metastatic lymph node discovered to be due to a GP in the fourth portion of the duodenum. These duodenal lesions are commonly confused with neuroendocrine neoplasms, which are of epithelial origin, and are distinguished from them using immunohistochemistry.
Keywords
Duodenal gangliocytic paraganglioma
Immunohistochemistry
Paraganglioma
Duodenal tumor

INTRODUCTION
Gangliocytic paraganglioma (GP) is an infrequent tumor occurring in the duodenum, with fewer than 300 reported cases globally.[1] Previous cases have commonly indicated its occurrence in the periampullary region.[2] This presentation details a case involving a young adult diagnosed with GP in the fourth part of the duodenum, associated with a metastatic lymph nodes. It is noteworthy that all paragangliomas (PGLs) have metastatic potential, and these duodenal lesions are often confused with neuroendocrine neoplasms (NENs) of epithelial origin, emphasizing the importance of immunohistochemistry (IHC) for accurate differentiation.[3]
CASE REPORT
A 30-year-old male presented with a localized upper abdominal pain, persisting for a month. Imaging revealed prominent lymph nodes near the para-aortic region. Subsequent upper gastrointestinal (GI) endoscopy was inconclusive.
The patient underwent positron emission tomography (PET) with fluorodeoxyglucose. It showed an enlarged node with increased metabolic activity (maximum standardized uptake value [SUVmax] ~ 16.38) seen at the para-aortic region of size 2.1 × 2.0 × 2.8 cm, adjacent to a focal enhancing submucosal lesion with a mild increase in metabolic activity at the level of the duodenojejunal flexure of size 1.0 × 0.6cm (SUVmax ~ 6.00) [Figure 1].
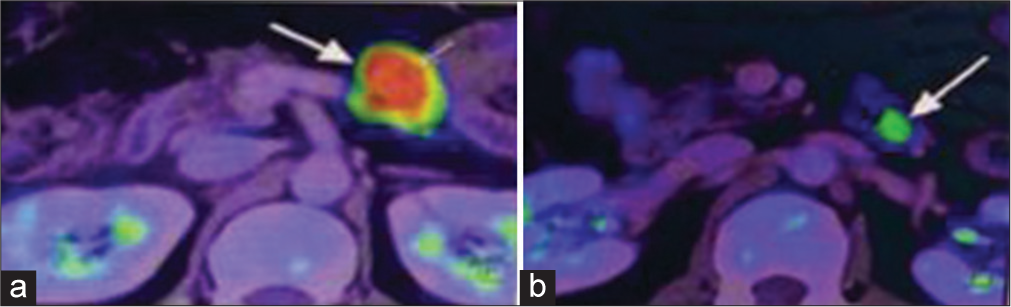
- (a) Enlarged lymph node identified on fluorodeoxyglucose-positron emission tomography computed tomography (FDG-PET CT) as shown by white arrow, (b) Submucosal lesion detected on FDG-PET CT as shown by white arrow.
Laparotomy was performed, and the lymph node was sent for biopsy [Figure 2]. The submucosal lesion was noted to be 1 × 1 cm in the fourth part of the duodenum.

- Enlarged lymph node identified intraoperatively.
The final histopathology was reported as GP, which showed solid nests, trabeculae, and tubular patterns. There were epithelioid cells that were polygonal to columnar with bland oval nuclei with stippled chromatin and pale eosinophilic, fine granular cytoplasm. There were scattered ganglion cells with large vesicular nuclei and prominent nucleoli. There were bland spindle cells forming broad fascicles enveloping the epithelioid and ganglion cells [Figure 3]. IHC showed positive for synaptophysin and chromogranin, weakly positive for panCK, and negative for inhibin and ER G. The scattered ganglion cells were positive for S100 [Figure 4]. The Ki67 score was 2%, suggesting it to be a neuroendocrine tumor (NET) grade 1 as per the 2020 World Health Organization classification of GI NETs.[4]
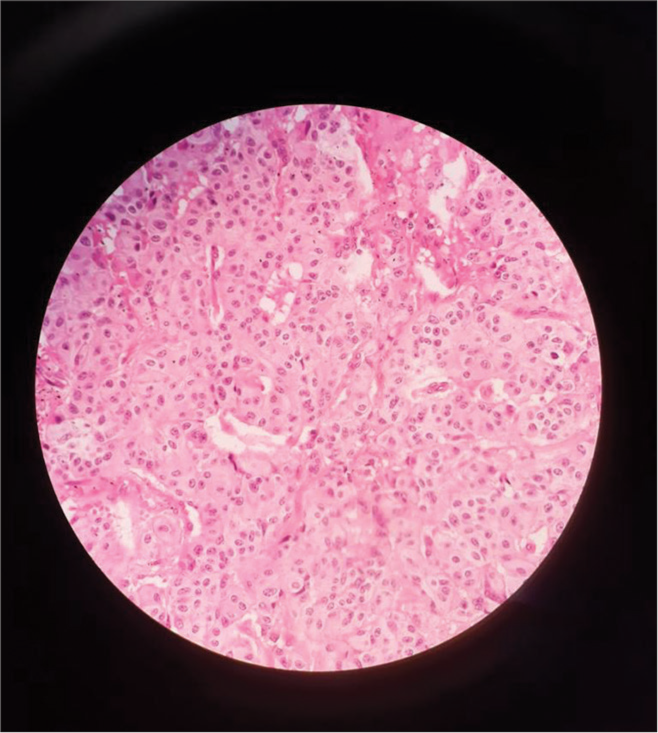
- Tumor cells nests as seen under the microscope
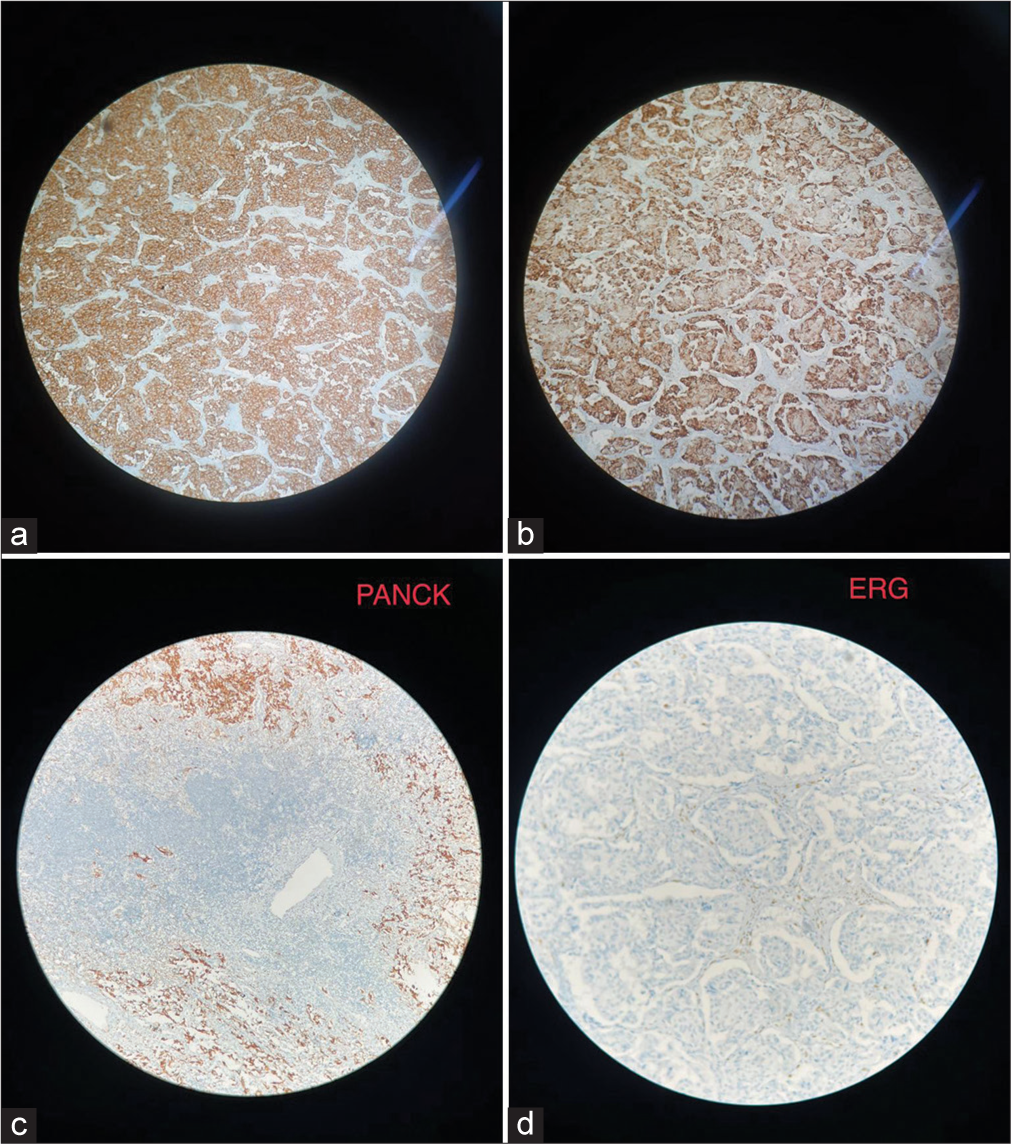
- (a) Positive for synaptophysin, (b) Positive for chromogranin, (c) Immunohistochemistry (IHC) image for Pan cytokeratin (PAN CK), (d) IHC image for ETS related gene (ERG).
A multidisciplinary team opinion was obtained, and he was planned for observation and follow-up. A baseline 68Ga DOTANOC positron emission tomography (PET) computed tomography scan was done, which showed a mild somatostatin receptor (SSTR) expressing primary lesion and a poor to mild SSTR expressing residual lymph nodal metastasis. The NET PET score was P4b/P5.[5]
The patient is currently on follow-up for observation and to watch for the progression of the tumor.
DISCUSSION
PGLs originate from chromaffin cells outside the adrenal gland and can occur along the autonomic nervous system ganglia.[6] PGLs originate from sympathetic or parasympathetic ganglia in the abdomen, thorax, and pelvis. PGLs may also originate from parasympathetic ganglia at the base of the skull and along the glossopharyngeal nerve and vagus nerve in the neck.[7] Intra-abdominal PGLs have an incidence of 1 in 500000.[8] GPs, a rare form of PGLs, are considered to originate from the pancreatic islet. These tumors pose a diagnostic challenge pre-operatively as these submucosal lesions are difficult to biopsy. However, when these tumors occur as polypoidal lesions, they can be managed endoscopically.[9]
These tumors can commonly be confused with low-grade NETs of the small bowel. As in the systematic review concluded by Okubo et al., these tumors have a male preponderance and are commonly localized to the duodenum. These tumors are considered to have a better prognosis than NETs.[10]
The demonstration of all three cells on examination and IHC helps to distinguish these tumors of the midgut.[3] Common differentials include gastrointestinal stromal tumor, schwannoma, and leiomyoma, which are distinguished by IHC.[10]
These tumors have a tendency to metastasize to the local draining lymph node bed, depending on the depth of invasion of the primary tumor. These tumors tend to have a benign course and rarely recur after resection with negative margins.[2]
Treatment protocols for GP are not well established due to the rarity of the tumor.
A recent literature review of 263 cases suggests that primary excision of the duodenal tumor is sufficient for disease control, as recurrence is rare in this tumor.[10] The rarity of this tumor in the fourth part of the duodenum is to be noted. Hence, in our patient, due to the small size of the tumor, the young age of the patient, and the consensus of the multidisciplinary team, a trial of observation and follow-up is underway.
CONCLUSION
Immunohistochemistry plays a key role in identification of the origin of the tumor. A multidisciplinary approach is vital in rare tumors due to the lack of established management protocols.
Acknowledgment
I thank my Professors, Dr.Balaji Singh, Dr.Raja Senthil, and Dr.Chandru (Associate professor), and Professor Dr.Sandhya and Dr.Pavithra (Associate professor) of the Department of Pathology, without whom this case report would not have materialized.
Ethical approval
The Institutional Review Board approval is not required.
Declaration of patient consent
Patient’s consent is not required as patients identity is not disclosed or compromised.
Conflicts of interest
There are no conflicts of interest.
Use of artificial intelligence (AI)-assisted technology for manuscript preparation
The authors confirm that there was no use of artificial intelligence (AI)-assisted technology for assisting in the writing or editing of the manuscript and no images were manipulated using AI.
Financial support and sponsorship
Nil.
References
- Duodenal gangliocytic paragangliomas-case series and literature review. Life. 2023;13:597.
- [CrossRef] [PubMed] [Google Scholar]
- Duodenal rare neuroendocrine tumor: Clinicopathological characteristics of patients with gangliocytic paraganglioma. Gastroenterol Res Pract. 2016;2016:5257312.
- [CrossRef] [PubMed] [Google Scholar]
- Duodenal gangliocytic paraganglioma: A case report and literature review. Int J Surg Case Rep. 2015;8:5-8.
- [CrossRef] [PubMed] [Google Scholar]
- The ENETS/WHO grading system for neuroendocrine neoplasms of the gastroenteropancreatic system: A review of the current state, limitations and proposals for modifications. Int J Endocr Oncol. 2016;3:203-19.
- [CrossRef] [PubMed] [Google Scholar]
- The NETPET score: Combining FDG and somatostatin receptor imaging for optimal management of patients with metastatic well-differentiated neuroendocrine tumors. Theranostics. 2017;7:1159-63.
- [CrossRef] [PubMed] [Google Scholar]
- Pheochromocytoma and paraganglioma: From epidemiology to clinical findings. Sisli Etfal Hastan Tip Bul. 2020;54:159-68.
- [CrossRef] [PubMed] [Google Scholar]
- Part 1 of 2: Advances in pathogenesis and diagnosis of pheochromocytoma and paraganglioma. Ann Surg Oncol. 2020;27:1329-37.
- [CrossRef] [PubMed] [Google Scholar]
- Primary intra-abdominal paraganglioma: A case report. World J Clin Cases. 2023;11:2276-81.
- [CrossRef] [PubMed] [Google Scholar]
- Is gangliocytic paraganglioma designated as a subtype of composite paragangliomas and originated from pancreas islet? A case report and review of literature. Front Endocrinol. 2022;13:847632.
- [CrossRef] [PubMed] [Google Scholar]
- Diagnosis, pathological findings, and clinical management of gangliocytic paraganglioma: A systematic review. Front Oncol. 2018;8:291.
- [CrossRef] [PubMed] [Google Scholar]




