
Translate this page into:
A rare presentation of kikuchi disease as hemophagocytic lymphohistiocytosis
*Corresponding author: Harish Kasarabada, Department of Internal Medicine, Army Hospital Research and Referral, Delhi, India. kasarabadaharish@gmail.com
-
Received: ,
Accepted: ,
How to cite this article: Kasarabada H, Singhal M, Singh D, Iyengar S, Joshi S. A rare presentation of kikuchi disease as hemophagocytic lymphohistiocytosis. Sri Ramachandra J Health Sci. 2023;3:72-5. doi: 10.25259/SRJHS_24_2023
Abstract
This case report deals with a young lady in her late 20’s who presented as a case of pyrexia of unknown origin complicated by the rapid development of encephalitis transaminitis and pancytopenia within a span of 24 h and later succumbed to her illness. Her clinical diagnosis and cause of death were hemophagocytic lymphohistiocytosis (HLH) secondary to neutropenic sepsis; however, an autopsy was recommended to ascertain the exact cause of death. Her autopsy examination revealed numerous necrotic enlarged lymph nodes with elevated lactate dehydrogenase levels in peritoneal fluid. The final diagnosis was multiorgan failure due to HLH in the case of Kikuchi disease. Kikuchi disease is usually a benign self-limiting illness presenting most commonly as cervical or axillary lymphadenopathy. This case report highlights a fatal case of Kikuchi disease due to HLH, which is an extremely rare entity.
Keywords
Kikuchi disease
Hemophagocytic lymphohistiocytosis
pyrexia of unknown origin

INTRODUCTION
Kikuchi disease, also called Kikuchi-Fujimoto disease or Kikuchi histiocytic necrotizing lymphadenitis, was originally described in young women and is a rare, benign condition of unknown cause usually characterized by cervical lymphadenopathy and fever. The disease is triggered by many inciting agents such as Epstein–Barr virus, herpes virus, and parvovirus.[1-3] Among various inciting agents, parvovirus B-19 showed an association of Kikuchi disease with hemophagocytic lymphohistiocytosis (HLH).[3] The principal mechanism of cellular destruction is apoptotic cell death mediated by cytotoxic cluster differentiation molecule 8 (CD8) T lymphocytes (CD8), along with histiocytes acting as enhancers.[4] The mean age at presentation in the USA was 30 years, along with female predisposition and there is no specific ethnic predisposition, although first described in Japan.[5] The most common clinical presentation is lymphadenopathy, followed by fever, rash, and arthralgia. Cervical and axillary groups of lymph nodes are the most common among affected lymph nodes.[6] Neurological involvement although rare, at times, may be complicated with aseptic meningitis, encephalitis, and ataxia.[7] HLH development in Kikuchi disease is rare, barring a few case reports.[8] Abnormal laboratory parameters may include leukopenia with the predominance of macrophages in bone marrow. The diagnosis of Kikuchi disease is made by an excision biopsy of the lymph node suggestive of necrotic lymphadenitis. Important differentials include tubercular lymphadenitis, lymphogranuloma venereum, systemic lupus erythematosus (SLE), Kawasaki disease, and lymphoma.[9] There is no effective treatment for Kikuchi disease, and it is a self-limiting disease that usually resolves within four weeks. Although high-dose glucocorticoids or in combination with intravenous immunoglobulin (IVIg), at times showed benefit.[10] HLH is an aggressive, life-threatening syndrome of abnormal and excess immune activation leading tissue destruction followed by organ failure. The hyperinflammatory or dysregulated immune state is thought to be caused by the absence of normal downregulation by activated macrophages and lymphocytes.[11] The most common clinical features include fever, bicytopenia, and splenomegaly. Abnormal parameters include transaminitis, hypertriglyceridemia, and high ferritin values. There is documented evidence of low or absent natural killer (NK) cell activity along with elevation of CD25 levels. Kikuchi disease-induced HLH is a rare entity, and when it happens it can lead to fatal outcomes. Here, in this case report, we discuss a similar fatal outcome in the case of Kikuchi disease, leading to HLH followed by organ failure and death.
CASE REPORT
A young lady in her late 20’s with no prior comorbidities presented to OPD (outpatient clinic) with a history of fever of seven-day duration. She describes her fever as continuous with no systemic associations, and she denies any history of cough, dyspnea, pain in the abdomen, lower urinary tract symptoms, headache, chest pain, and palpitations. She also denies a history of arthralgia, rash, and hair loss at the time of presentation and in the past too. She is married with one healthy male child who is three years old, and her obstetric history was uneventful with normal menstrual cycles. Her preliminary investigations included complete blood counts, urine microscopy, biochemical parameters, tropical fever workup, Widal test, chest X-ray, and ultrasound abdomen. Among these, the only notable abnormality was leukopenia, with an absolute neutrophil count of around 1500 cells/cubic millimeter and a modest elevation of liver transaminases. She was admitted with a case of pyrexia of unknown origin (PUO) and was managed with broad-spectrum antibiotics and the best supportive therapy. She was then subjected to many investigations, such as echocardiography, acute phase reactants, and fundoscopy. All the reports were within normal limits; following 12 h of her admission, she developed two episodes of generalized tonic-clonic seizure. She underwent emergency neuroimaging of the brain, which was normal, followed by cerebrospinal fluid analysis, which did not reveal any abnormality. She had a rapid downhill course within a span of 12 h, developing shock, developing pancytopenia, worsening transaminitis, elevated acute phase reactants (ferritin > 1000 mcg/L), and hypertriglyceridemia requiring organ support (mechanical ventilation and vasopressors). A clinical diagnosis of HLH was made, and the lady was started on high-dose methylprednisolone along IVIg along with broad-spectrum antibiotics, antivirals, and antiepileptic drugs. However, she was refractory to treatment, following which she succumbed to her illness. She was advised to autopsy to ascertain the exact cause of death.
Follow-up
Her autopsy report’s salient findings were
Intra-abdominal necrotizing lymphadenitis with high lactate dehydrogenase in peritoneal fluid following necrotizing lymphadenitis as shown in Figures 1-3 which features hemophagocytic, histiocytic activity along with necrosis.
Diffuse bilateral alveolar damage.
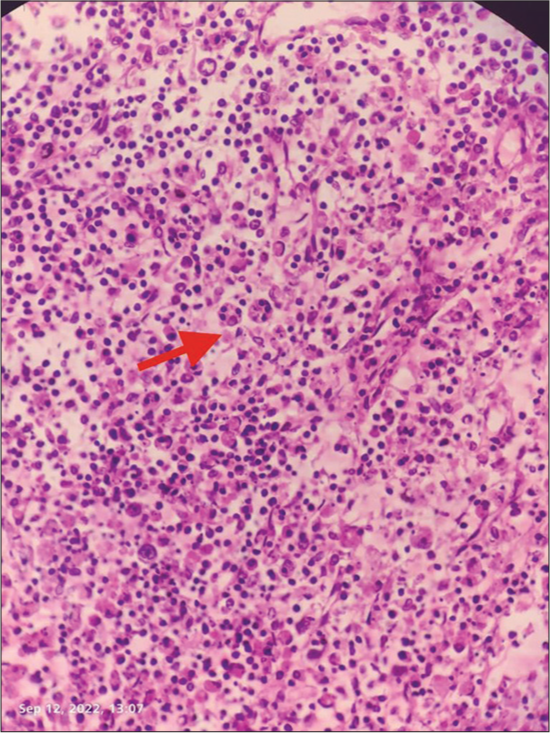
- Evidence of hemp phagocytosis in lymph node biopsy (as shown below by arrow).
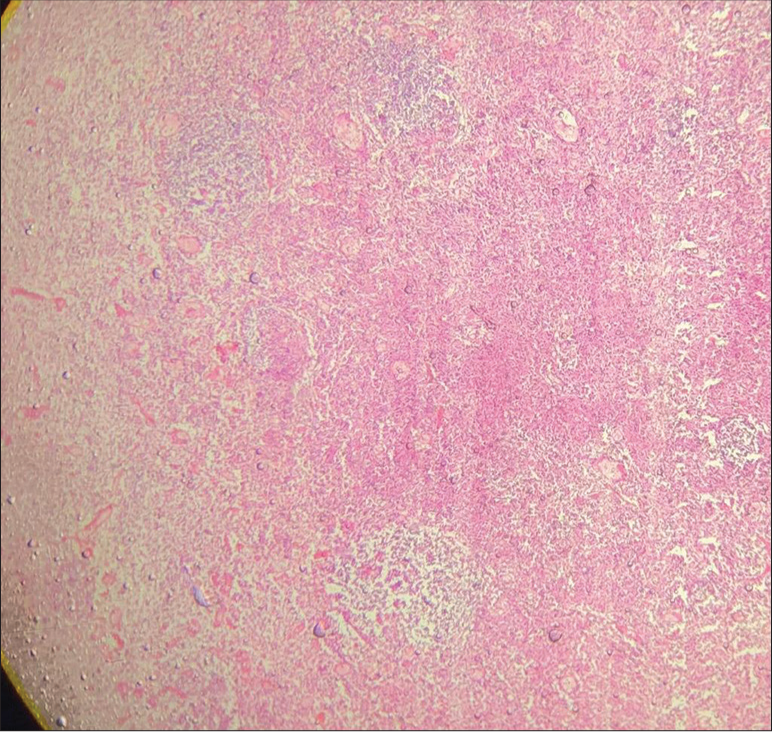
- Low-power view shows confluent necrosis with partial preservation of the lymph node architecture in ×5 magnification.
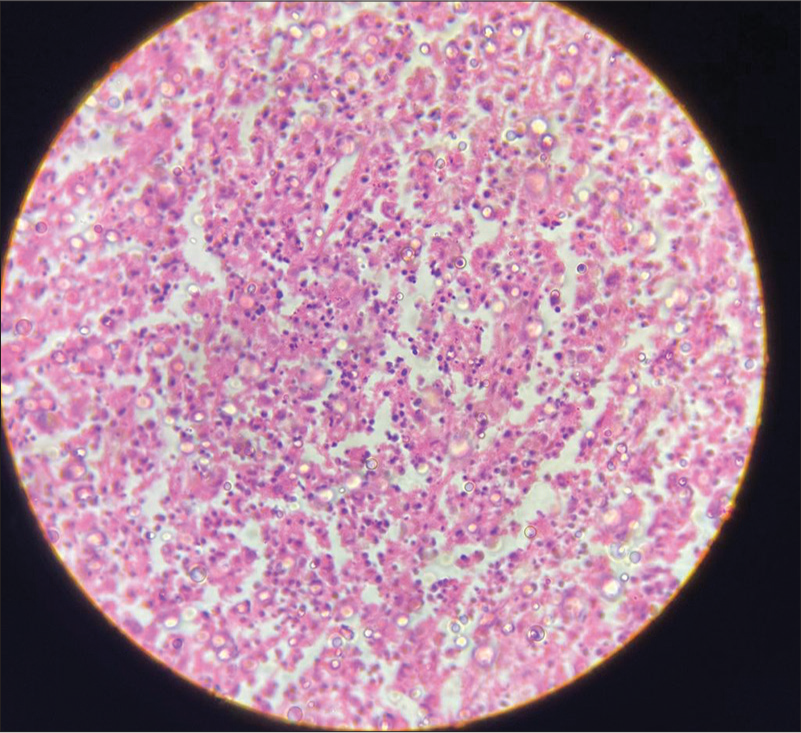
- High-power view shows apoptotic cells with nuclear debris, as well as admixed histiocytes and transformed lymphocytes in ×40 magnification.
The cause of death was attributed to multiorgan failure due to HLH in the case of Kikuchi disease.
DISCUSSION
Kikuchi disease presenting as a case of PUO is very rare, and it usually presents as a benign self-limiting illness characterized by cervical or axillary lymphadenopathy along with constitutional symptoms such as fever and arthralgia. Earlier described in Japan, it has a worldwide distribution with female predisposition occurring commonly in the adolescent age group. HLH can be described as primary, which has genetic susceptibility and secondary or HLH syndrome, which is secondary to other conditions such as sepsis, SLE, or Kikuchi disease. HLH presents with pancytopenia, elevated acute phase reactants, and hypertriglyceridemia with low or absent NK cell activity. It is usually fatal and requires management with a high dose of immunosuppression or chemotherapy. Here, there is a rare case where a lady presented with febrile illness of 1-week duration with a probable early potential clue of leukopenia as a part of impending HLH. From this case report, retrospectively, we can understand that Kikuchi disease can progress rapidly and lead to secondary HLH. However, the pathogenesis of Kikuchi disease progressing to HLH remains unclear.[12] Duan et al. study and literature review suggested that HLH associated with Kikuchi disease occurs most commonly during childhood and may have a less aggressive clinical course and better prognosis than in adults, but sometimes, it may still be fatal.[13] There is not much literature on the presentation of Kikuchi disease as HLH in adults, barring a few case reports. The lady had a rapid downhill course within a span of 12 h despite early provisional prediction of HLH and immediate medical intervention with high-dose corticosteroids and IVIg. However, a study conducted by Trottestam et al. suggested that treatment regimens for HLH include high-dose corticosteroids or a combination of corticosteroids and IVIg and, in refractory cases, a combination of dexamethasone and etoposide for induction, followed by maintenance either with etoposide or dexamethasone.[12] We understand that HLH is a life-threatening condition, but early treatment with etoposide would have benefitted in this patient is yet to be clear. We understand from this case that Kikuchi disease, which usually presents as cervical and axillary lymphadenopathy, can also present with abdominal lymphadenopathy. As we see in this case report, the clinical diagnosis is HLH and Kikuchi disease was an incidental finding on autopsy and diagnosis confirmed after autopsy, the enlarged necrotic abdominal lymph nodes did not result in any related symptoms making it even more difficult to localize to a specific system. Hereby, we conclude by stating that the diagnosis of Kikuchi disease was extremely challenging, and with this experience, we shall consider the diagnosis of Kikuchi disease as a part of differentials in a young lady presenting with PUO in the future and finally state that HLH can result secondary from Kikuchi disease and is as an extremely rare entity.
CONCLUSION
We conclude that this case report highlights a rare association between Kikuchi disease and HLH with involvement of intra-abdominal lymph nodes. It was an extremely difficult diagnosis to arrive from a case of PUO in view of its non-specific presentation of symptoms. In view of the same in future we would consider Kikuchi disease as one among the list of differentials in a young lady with PUO, leucopenia with non specific abdominal pain.
A rare association of HLH and Kikuchi disease.
Rare involvement of abdominal lymph nodes secondary to Kikuchi disease.
A young lady with PUO and leukopenia, Kikuchi is considered as one among the list of differentials.
Ethical approval
The research/study complied with the Helsinki Declaration of 1964.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent.
Conflicts of interest
There are no conflicts of interest.
Use of artificial intelligence (AI)-assisted technology for manuscript preparation
The authors confirm that there was no use of artificial intelligence (AI)-assisted technology for assisting in the writing or editing of the manuscript and no images were manipulated using AI.
Financial support and sponsorship
Nil.
References
- EBV-associated Kikuchi's histiocytic necrotizing lymphadenitis with cutaneous manifestations. J Am Acad Dermatol. 1997;36:342-6.
- [CrossRef] [PubMed] [Google Scholar]
- Detection of human herpesvirus DNA in Kikuchi-Fujimoto disease and reactive lymphoid hyperplasia. Int J Clin Exp Pathol. 2008;1:362-8.
- [Google Scholar]
- Parvovirus B19-associated haemophagocytic syndrome with lymphadenopathy resembling histiocytic necrotizing lymphadenitis (Kikuchi's disease) Br J Haematol. 1997;96:868-71.
- [CrossRef] [PubMed] [Google Scholar]
- Apoptotic cell death in Kikuchi's disease: A TEM study. Acta Otolaryngol Suppl. 1998;538:250-3.
- [CrossRef] [Google Scholar]
- Kikuchi's histiocytic necrotizing lymphadenitis: An analysis of 108 cases with emphasis on differential diagnosis. Semin Diagn Pathol. 1988;5:329-45.
- [Google Scholar]
- Kikuchi-Fujimoto disease: Analysis of 244 cases. Clin Rheumatol. 2007;26:50-4.
- [CrossRef] [PubMed] [Google Scholar]
- Histiocytic necrotizing lymphadenitis (Kikuchi's disease) with aseptic meningitis. J Neurol Sci. 1999;163:187-91.
- [CrossRef] [PubMed] [Google Scholar]
- A case of haemophagocytic syndrome and Kikuchi-Fujimoto disease occurring concurrently in a 17-year-old female. Int J Clin Pract. 2000;54:547-9.
- [CrossRef] [PubMed] [Google Scholar]
- Severe Kikuchi's disease responsive to immune modulation. Singapore Med J. 2010;51:e18-21.
- [Google Scholar]
- Histiocytic disorders: Recent insights into pathophysiology and practical guidelines. Biol Blood Marrow Transplant. 2010;16(1 Suppl):S82-9.
- [CrossRef] [PubMed] [Google Scholar]
- Chemoimmunotherapy for hemophagocytic lymphohistiocytosis: Long-term results of the HLH-94 treatment protocol. Blood. 2011;118:4577-84.
- [CrossRef] [PubMed] [Google Scholar]
- Kikuchi's disease with hemophagocytic lymphohistiocytosis: A case report and literature review. Medicine (Baltimore). 2020;99:e23500.
- [CrossRef] [PubMed] [Google Scholar]




